3. PIPELINE MODE TUTORIAL
The pipeline mode of VariantSeq allows you to execute all steps of a protocol automatically as a pipeline. To perform the tutorial in pipeline mode, click on the “VariantSeq protocols” tab in the Top menu and select the option Pipeline Mode. A list will appear to show the available pipelines and filters based on types of reads, protocol mode, etc (Fig. 5). You only need to apply the filters and click run. This will pass you to the next section, which is a set of nested interfaces to:
1. Upload the input files and Refseq needed by the pipeline (i.e., fastq files, reference genome, PON and Training, and truth sets).
2. Declare the output folder to deposit the output results.
3. Declare the experiment design (samples to analyze etc).
4. Configure the parameters and options for each tool used at each step (for example, “FastQC” for quality analysis, “PRINSEQ” for preprocessing, “BWA” for mapping, “Picard Tools and GATK” for postprocessing, and GATK for calling and filtering and VEP for annotation).
5. Run the pipeline.
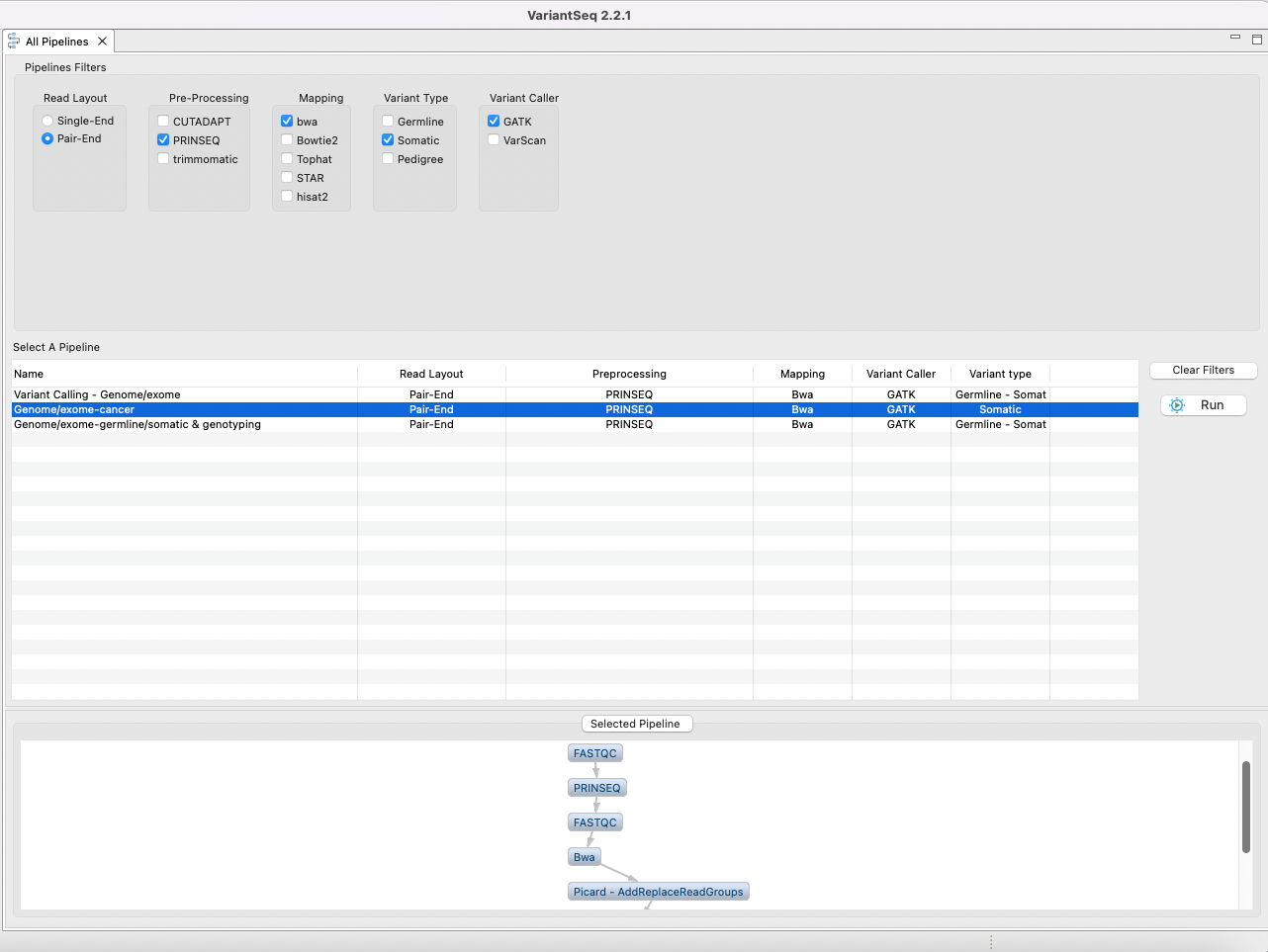
Figure 5. Pipeline configuration interface.
When you have the tutorial material ready, access the pipeline designer interface. VariantSeq Protocols → Pipeline mode and proceed as indicated in video 11.
Video 11. Protocol for SPMI analysis with VariantSeq using the pipeline mode
|
Expected results from SPMI protocol using the Pipeline execution mode: When the pipeline mode is complete, you will receive the differential expression results with the reads mapped against the reference genome. The expected results of this step are available in the following link Pipeline mode results You can check if the job was successfully completed by accessing the job tracking panel of VariantSeq. To learn more about any tool used in the pipeline and their outputs, see their respective manuals as indicated in previous sections of this tutorial. |
|---|
