3. PIPELINE MODE TUTORIAL
The pipeline mode of RNAseq allows you to execute all steps of a protocol automatically as a pipeline. The only exception is the GO enrichment step as this is not yet implemented in the pipeline mode. Thus, the pipeline mode currently finishes after the differential expression analysis. To perform the tutorial in pipeline mode, click on the tab “RNASeq protocols” on the Top menu and then select the option Pipeline Mode. A first graphical list of available pipelines will appear based on distinct combinations of tools (Figure 4). Besides, you can select several filters to list only the appropriate pipelines for an analysis according to different criteria including types of reads, protocol mode, etc. You only need to apply your filters and click run. In doing so, you will pass to the next section, which is a set of nested interfaces to:
1. Upload any input data or material needed by the pipeline (i.e., fastq files, reference genome or GTF/GFF file).
3. Declare the experiment design (groups to compare, samples belonging to each group, replicates, etc).
4. Configure the parameters and options of each tool used per step (for example, “FastQC” for quality analysis, “Trimmomatic” for preprocessing, “Tophat” for mapping, “Cufflinks” for transcriptome assembly and “Cuffdiff” for differential gene expression).
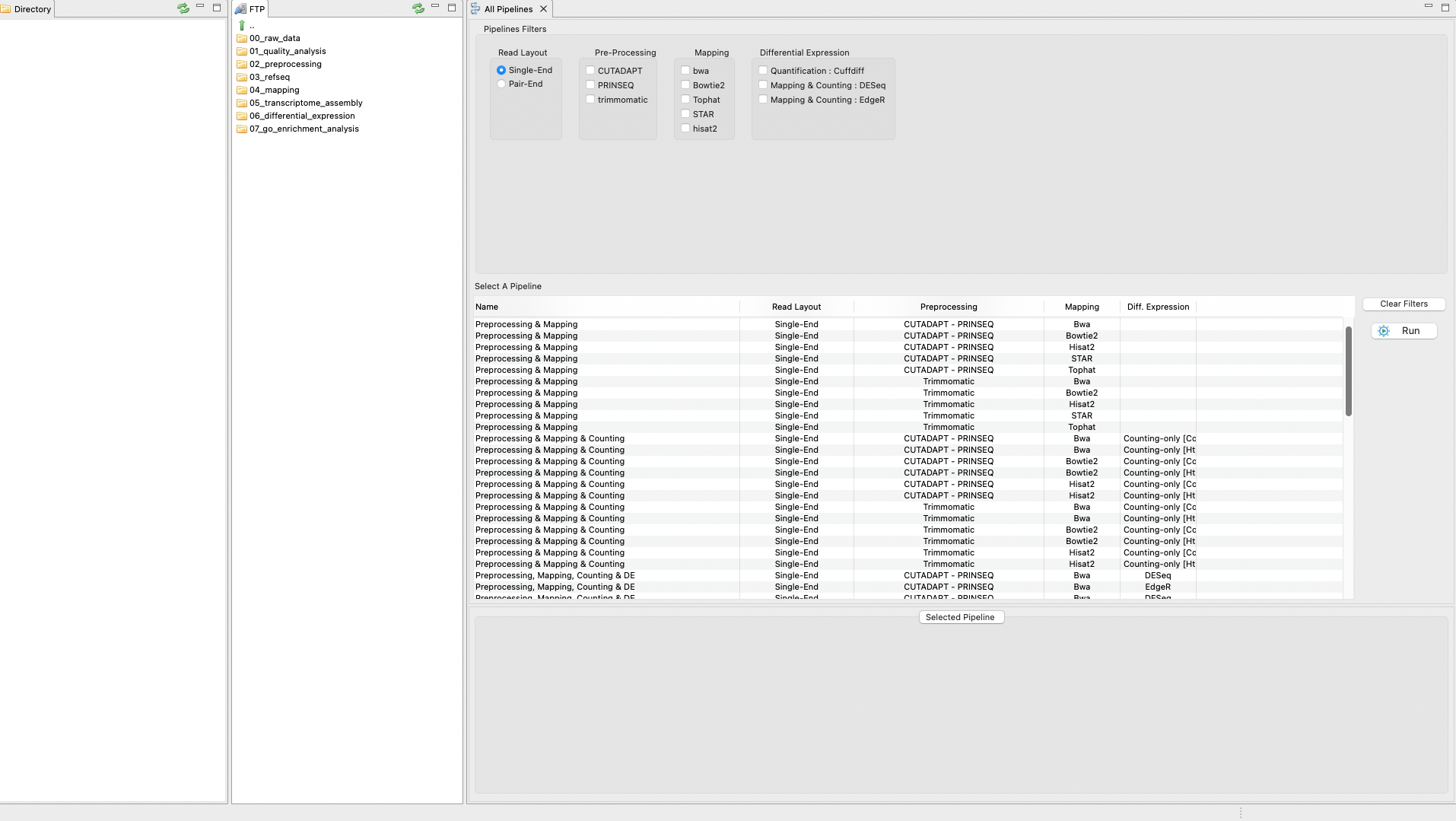
In this part of the tutorial, we reproduce both protocols (“TopHat/Hisat2 & Cufflinks” and “Mapping & Counting”) using the pipeline mode except for the step for the GOSeq analysis that is not currently included in the pipeline mode.
